A clonagem de
organismos multicelulares pode ser realizada de duas maneiras:
· Interferindo-se
em processos naturais de reprodução;
· Trabalhando-se
a partir de células somáticas que normalmente não atuam nos mecanismos
reprodutivos.
CLONAGEM
A PARTIR DE MECANISMOS NORMAIS DE REPRODUÇÃO
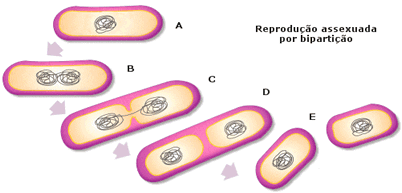
No
caso de plantas, o procedimento mais utilizado é a reprodução de indivíduos
idênticos por reprodução assexuada.
No
caso de animais, como por exemplo, os mamíferos estimulam-se em laboratório o
surgimento de gêmeos monozigóticos. Para isso, selecionam-se os animais que
apresentam características desejadas, faz-se a coleta de sêmen e ovócitos e
promove-se a fecundação no próprio laboratório.
Assim
que o zigoto se forma e que as primeiras divisões celulares se iniciam, as
células originadas são separadas artificialmente e implantadas em fêmeas (mães
de aluguel), onde de completa o desenvolvimento embrionário.
Cada uma
dessas células dará origem a um individuo geneticamente idêntico. Formam-se,
então clones de animais de interesse para o ser humano.
.jpg)
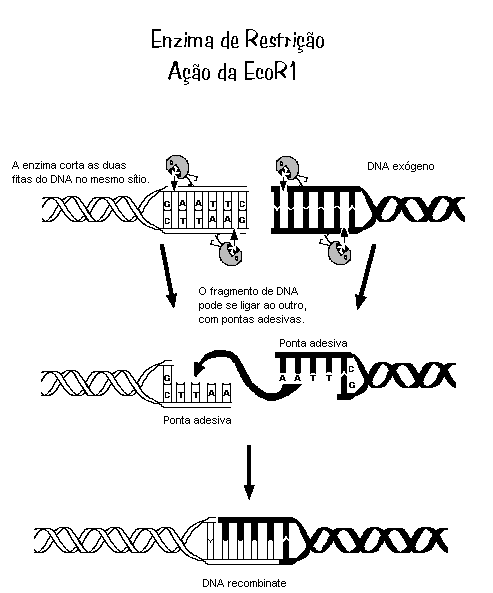
Um exemplo muito conhecido e divulgado de clonagem a partir
de células somáticas é o caso da ovelha Dolly (foi o
primeiro mamífero a ser clonado com sucesso a partir de
uma célula adulta.)
Nesse
caso, uma célula receptora, o ovócito retirado do ovário de uma ovelha da
raça blackface teve seu material genético removido com auxilio
de uma micropipeta. Uma célula (2n) retirada da glândula mamaria de uma ovelha
adulta da raça finn dorset foi mantida em condições que a
tornaram pouco ativa. Essa célula foi fundida ao ovócito desprovido de material
genético nuclear.
O
ovócito, agora com o núcleo 2n recebido da célula somática, foi estimulado a
iniciar o desenvolvimento embrionário.
TERAPIA
GÊNICA
Por terapia gênica se
entende a transferência de material genético com o propósito de prevenir ou
curar uma enfermidade qualquer. No caso de enfermidades genéticas, nas
quais um gene está defeituoso ou ausente, a terapia gênica consiste em
transferir a versão funcional do gene para o organismo portador da doença, de
modo a reparar o defeito. Se trata de uma idéia muito simples, mas como veremos
sua realização prática apresenta vários obstáculos.
Primeira etapa: o isolamento do gene.
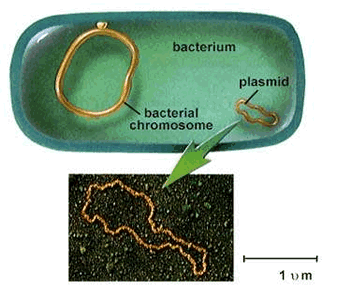
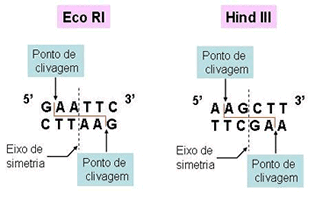
Um gene é
uma porção de DNA que contém a informação necessária para sintetizar uma
proteína. Transferir um gene é transferir um pedaço particular de DNA.
Portanto, é necessário antes de tudo, possuir “em mãos” o pedaço correto.
As enfermidades genéticas conhecidas estão ao redor de 5000, cada uma
causada por uma alteração genética diferente. O primeiro passo para a terapia
gênica é identificar o gene responsável pela enfermidade. Subsequentemente,
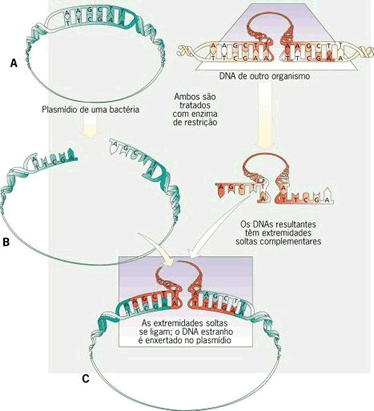
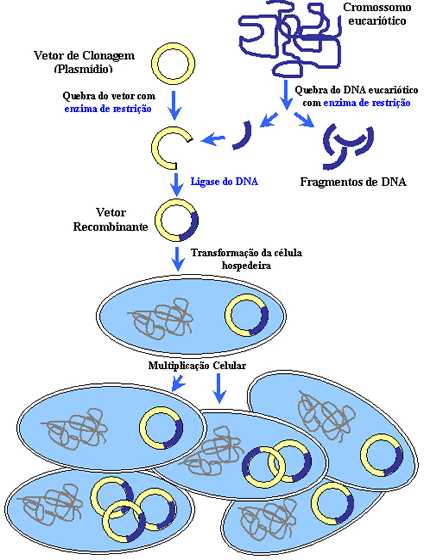
pelas técnicas de biologia molecular é possível adquirir um pedaço de DNA que contém
este gene. Esta primeira etapa é chamada de isolamento ou clonagem do
gene.
Qualquer enfermidade é candidata a terapia gênica, desde que o gene
esteja isolado para a transferência.
Graças ao progresso da biologia molecular esta primeira etapa é relativamente
simples em comparação a alguns anos atrás. Tem sido possível isolar numerosos
genes causadores de doenças genéticas e, se descobrem outros a cada
semana.
Estas
condições mostram qual é o objetivo da transferência gênica. Os procedimentos
da terapia gênica in vivo consistem em transferir o DNA
diretamente para as células ou para os tecidos do paciente.
Nos procedimentos ex-vivo, o DNA é primeiramente
transferido para células isoladas de um organismo, previamente crescidas em
laboratório. As células isoladas são assim modificadas e podem ser introduzidas
no paciente. Este método é indireto e mais demorado, porém oferece a vantagem
de uma eficiência melhor da transferência e a possibilidade de selecionar e
ampliar as células modificadas antes da reintrodução.
Como se transfere o DNA a célula hospedeira?
Os
procedimentos de transferência do DNA in vivo ou em ex-vivo têm
o mesmo propósito: o gene deve ser transferido para dentro das células, e uma
vez inserido tem que resistir bastante tempo. Neste tempo, o gene tem que
produzir grandes quantidades de proteína para reparar o defeito genético. Essas
características podem ser resumidas em um único conceito: o gene estranho
precisa se expressar de modo efetivo no organismo que o receberá.
O sistema
mais simples seria, naturalmente, injetar o DNA diretamente nas células ou
nos tecidos do organismo a ser tratado. Na prática, este sistema é
extremamente ineficaz: o DNA desnudo quase não apresenta efeito nas células.
Além disso, essa tentativa requer a injeção em uma única célula ou grupos de
células do paciente.
Por isto, quase todas as técnicas atuais para a transferência de
material genético implicam o uso de vetores, para transportar o DNA para
as células hospedeiras.
Os
vetores virais são vírus manipulados geneticamente, de modo a reduzir a sua
patogenicidade, sem anular totalmente o seu poder de infectar as células do
hospedeiro (leia mais sobre isso no conteúdo de vírus) Com as técnicas da
engenharia genética é possível somar ao DNA do vírus o gene que se quer
transferir a determinada célula. Deste modo, o vírus infectando a célula, trará
consigo uma ou mais cópias do gene desejado.
Os retrovírus possuem a habilidade de integrar o seu
DNA dentro dos cromossomos da célula infectada. Então, o gene será inserido no
genoma das células hospedeiras e, podem assim ser transmitidos a todas as
células-filhas das infectadas. Eles infectam somente as células que estão
proliferando.
Os lentivírus,
como o HIV, permitem também transferir material genético para células que não
proliferam (como os neurônios e células do fígado) ou para células refratárias
para o retrovírus (como as células retiradas da medula óssea).
Os adenoassociados de vírus também integram o seu DNA ao
cromossomo da célula hospedeira. Eles têm a vantagem de serem inofensivos para
a natureza em relação ao retrovírus, mas não são capazes de transportar genes
de dimensões grandes.
Os adenovírus não são capazes de integrar o seu DNA
ao cromossomo da célula hospedeira. Eles podem transportar genes de grandes
dimensões, mas a expressão deles não dura muito tempo.
Os vetores
não virais
Os lipossomos são essencialmente os únicos vetores
não virais utilizados freqüentemente. As esferas de lipídeos podem ser um
importante meio para a transferência gênica. Em comparação aos vírus, eles têm
a vantagem de não introduzir algum risco em condições de segurança, mas eles
não possuem grande eficiência e são muito seletivos.
Os limites da terapia gênica
As
principais dificuldades enfrentadas por pesquisadores que lidam com
terapia gênica são as seguintes:
A eficiência da
transferência
Um
especialista em terapia gênica uma vez afirmou: “a terapia gênica” sofre de
três problemas técnicos principais; a transferência, a transferência e a
transferência”.
Nos
estudos de terapia gênica, a maior parte dos esforços é concentrada na procura
de vetores que possam transferir o DNA de modo eficiente, principalmente para
células desejadas. Nestes últimos anos foram inventados e testados uma grande
variedade de vetores, alguns dos quais com chances de expressar o gene estranho
em um tipo celular específico (como glóbulos brancos, células do músculo, das
vias respiratórias, etc.). Alguns destes estão em vias de experimentação no
homem e, a esperança é que apareçam resultados bons.
A duração da expressão
A terapia
gênica é praticamente inútil se a expressão do gene estranho não é mantida por
um bom período de tempo. As pesquisas estão orientadas de maneira a desenvolver
sistemas que apresentem uma expressão duradoura, de modo a submeter o paciente a um
único tratamento, ou a tratamentos repetidos em períodos maiores (anos).
A segurança do procedimento
Este é um
problema particularmente evidente para os vetores virais. Alguns destes derivam
de vírus perigosos como
o HIV. É então necessário que antes do uso destes vetores sejam submetidos a
critérios de segurança, particularmente no que concerne a presença de genes que
podem determinar a patogenicidade do vírus utilizado para infectar (transferir
o gene desejado) para as células do hospedeiro.
A reação imunitária
Como toda
substância estranha, o produto do gene novo, o gene propriamente dito ou seu
vetor podem instigar uma resposta imunitária no organismo sob tratamento. Isto
pode causar a eliminação das células modificadas geneticamente, ou a inativação
da proteína produzida pelo gene novo. No desenvolvimento de estratégias novas
de terapia gênica procura-se evitar respostas imunitárias ao vetor ou ao
produto do gene introduzido. Se trata de uma tarefa difícil e freqüentemente
empírica, mas isso é cada vez mais usado em inovações no campo da imunologia.










.jpg){kind=link}